

FRAGMENT REPLACEMENT & MOLECULAR DESIGN
BROOD
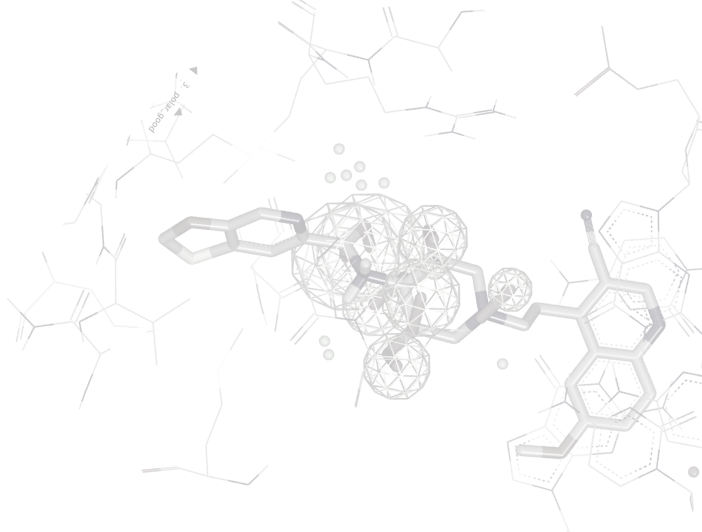
BROOD is a generative chemistry program designed to help explore chemical and property space around a hit or lead molecule. BROOD generates analogs of the lead by replacing selected fragments in the molecule with fragments that have similar shape and electrostatics, yet with selectively modified molecular properties.
BROOD fragment searching has multiple applications, including lead-hopping, side-chain enumeration, patent breaking, fragment merging, property manipulation, and patent protection by SAR expansion.
Using OpenEye’s BROOD software for scaffold hopping, we successfully discovered a new chemical series from our lead compound, with equipotency and improved stability. – Laurent D., Lundbeck
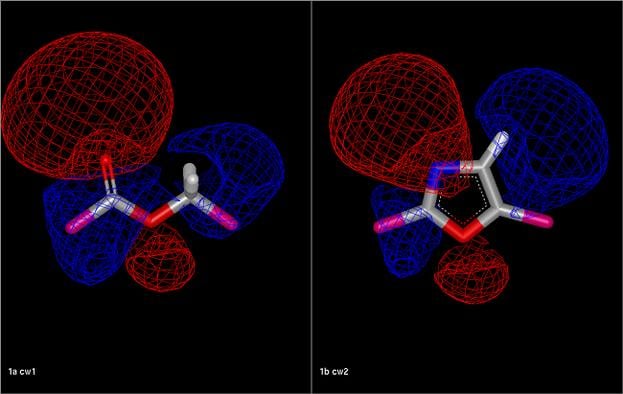
Comparison of an ester fragment and an oxazole fragment showing the electrostatic isopotential contour surfaces. The electrostatic Tanimoto between the two fragments is 0.54.

