

Iridium Database
The accuracy and precision of any prediction can never be better than the accuracy and precision of the data used to generate the model making the prediction.
In spite of an enormous investment, the true promise of structure-based design has largely eluded researchers for the past three decades. There are many reasons why this promise has not been fulfilled. The primary motivation behind Iridium is to address the problem of the protein-ligand structural databases used to develop and validate structure-based design tools, in particular, docking algorithms.
The accuracy and precision of any prediction can never be better than the accuracy and precision of the data used to generate the model making the prediction. However, determining the accuracy and precision of experimental data is not a simple task and as a result, it has been neglected in a number of cases.
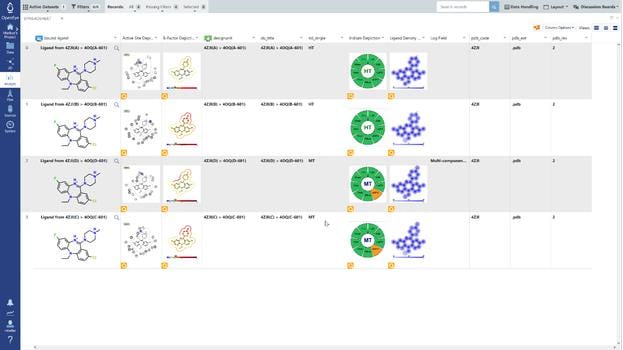
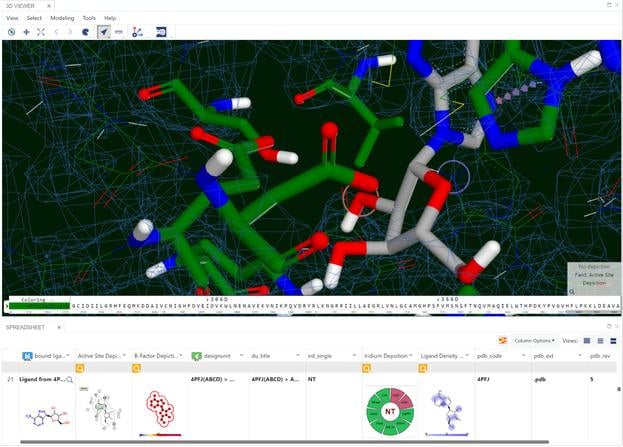
A set of 728 structures, taken from four published receptor-ligand structural databases, were analyzed to determine the quality of the structures. From this set 495 structures were rejected because the experimental data (electron density) for those structures was not publically available. The ligands from the resulting 233 structures were compared with primary and secondary literature and where discrepancies existed, they were curated to ensure that atom identity, functional groups, bond orders, atom charges, stereochemistry and tautomer states matched between the literature and the representation derived from the Protein Data Bank (PDB) file. Of the 233 structures analyzed, 47 (20.2%) required curation. In addition to ligand curation, binding affinity data was collected for 85.8% of the structures. The curated structures were then re-refined. The resulting structures were checked for quality using a number of criteria. The application of these criteria resulted in a receptor-ligand data set of 121 structures named Iridium-HT for Highly Trustworthy. Another 104 structures that violated some of the quality criteria were named Iridium-MT for Moderately Trustworthy. Eight structures had particularly pernicious problems and were named Iridium-NT for Not Trustworthy. A prerequisite for high-quality predictions in structure-based design is high-quality receptor-ligand structures. Iridium-HT provides such structures. Drug Disc. Today, 12, 1270 (2012)
Iridium is freely available and may be downloaded at the link below.
Download Iridium Database
