[WidgetDefinition{typeName=module, name=additional_post_content, label=Additional Post Content, isOverrideable=true, isGlobal=false, module_id=150680906540}, WidgetDefinition{typeName=module, name=custom_settings, label=Global - Custom Setting, isOverrideable=true, isGlobal=false, module_id=92467829120}]
OpenEye Scientific is now part of Cadence
Solutions
OpenEye Solutions
Science | Speed | Scale
Learn More
Product
Orion® Molecular Design Platform
Small Molecule Discovery Suite
Antibody Discovery Suite
Formulations Suite
Gaussian Module
Applications
Cheminformatics & Modeling Toolkits
Visualization & Data Management
Curated Databases
Topic
Virtual Screening
Lead Discovery
Lead Optimization
Drug Formulation
Antibody Discovery
Structural Biology
Interest
Ligand-Based Design
Structure-Based Design
Properties Calculations
Antibody Engineering
Biomolecular Modeling
Protein & Data Preparation
Crystal Structure Prediction
Quantum Chemistry
Machine Learning
Artificial Intelligence
Industry Sector
Biotech & Pharmaceuticals
Academic Institutions
Agrochemicals
Flavors & Fragrances
Services
OpenEye Services
Find out more about our services
Services
Drug Discovery Consulting
Crystal Structure Prediction for Drug Formulation
Custom Scientific Method Development
Partnerships and Engagements
Resources
OpenEye Resources
Find out more about our resources
Resources
Events
News
Documentation
Presentations
Webinars
Publications
Download
Support
Academic Licensing
Software Integration Partners
About
About OpenEye
Find out more about OpenEye
About
Why OpenEye
Team
Scientific Advisory Board
Careers
Let's Connect
Let's Connect
Solutions
OpenEye Solutions
Science | Speed | Scale
Learn More
Product
Orion® Molecular Design Platform
Small Molecule Discovery Suite
Antibody Discovery Suite
Formulations Suite
Gaussian Module
Applications
Cheminformatics & Modeling Toolkits
Visualization & Data Management
Curated Databases
Topic
Virtual Screening
Lead Discovery
Lead Optimization
Drug Formulation
Antibody Discovery
Structural Biology
Interest
Ligand-Based Design
Structure-Based Design
Properties Calculations
Antibody Engineering
Biomolecular Modeling
Protein & Data Preparation
Crystal Structure Prediction
Quantum Chemistry
Machine Learning
Artificial Intelligence
Industry Sector
Biotech & Pharmaceuticals
Academic Institutions
Agrochemicals
Flavors & Fragrances
Services
OpenEye Services
Find out more about our services
Services
Drug Discovery Consulting
Crystal Structure Prediction for Drug Formulation
Custom Scientific Method Development
Partnerships and Engagements
Resources
OpenEye Resources
Find out more about our resources
Resources
Events
News
Documentation
Presentations
Webinars
Publications
Download
Support
Academic Licensing
Software Integration Partners
About
About OpenEye
Find out more about OpenEye
About
Why OpenEye
Team
Scientific Advisory Board
Careers
Blog
Subscribe to our blog
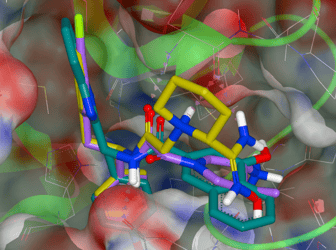
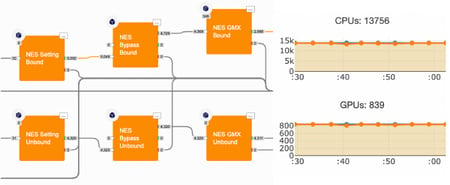
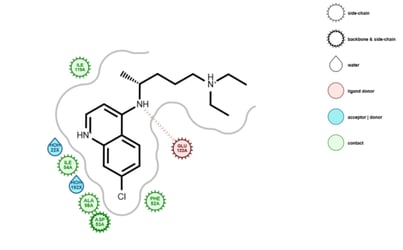
Relative Binding Free Energy with Non-Equilibrium Switching in Orion
By Gaetano Calabro PhD, OpenEye & Christopher Bayly, PhD OpenEye A variety of free energy simulation methods, such as FEP, TI, and λ dynamics, make use of atomistic MD or …
Accelerating Similarity and Substructure Searches
The number of drug-like molecules that could exist is staggering. Estimates range as high as 1060, more than the number of atoms in the Universe. Commercially available …
Browse by topic
"
business
COVID-19
Giga docking
GraphSim TK
large scale virtual screening
licensing
News
OEChem TK
OpenEye
Python
ROCS
All
GraphSim TK
News
OEChem TK
business
COVID-19
Giga docking
GraphSim TK
large scale virtual screening
licensing
News
OEChem TK
OpenEye
Python
ROCS
OpenEye Releases Additional Virtual Screening COVID-19 Data to Public
Read Post
Accelerating Similarity and Substructure Searches
Read Post
OpenEye deploys the Orion platform to find COVID-19 therapeutics
Read Post
Relative Binding Free Energy with Non-Equilibrium Switching in Orion
Read Post
Easier is Better
Read Post
Using Apache Maven with OpenEye
Read Post
Grapheme: Advancing Protein-Ligand Visualization
Read Post
Our documentation has moved
Read Post
Recommendation System for Compound Selection
Read Post