SZYBKI [1] optimizes molecular structures with the force field, either with or without solvent effect, to yield quality 3D molecular structures for use as input to other programs. Since the chemistry of molecular interactions is a matter of shape and electrostatics, it is impossible to consider either without reasonable 3D molecular structures.
SZYBKI also refines portions of a protein structure and optimize ligands within a protein active site, making it useful in conjunction with docking programs.
Supported force fields in SZYBKI includes OpenFF (Open Force Field Initiative) force fields Sage and Parsley, AMBER Protein force fields ff14SB and AMBER99, and the MMFF94 (Merck Molecular Force Field).
Freeform is a utility program included with SZYBKI that provides two functionalities (1) the solvation energy of the free ligand, (2) the free energy of going from an ensemble of solution phase conformers to a single, bioactive conformation.
For more detailed information on SZYBKI, check out the links below:
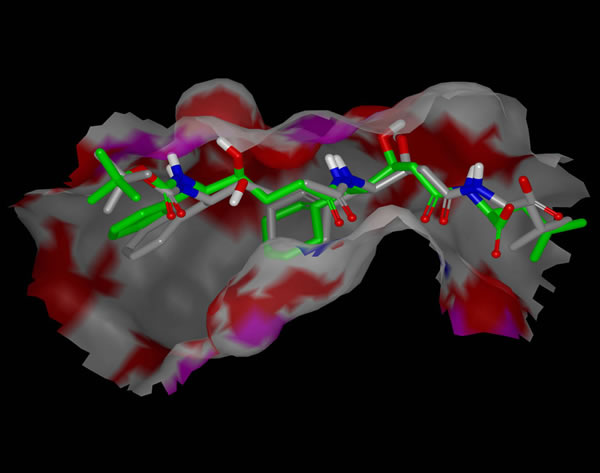
In situ optimization of a ligand with SZYBKI.
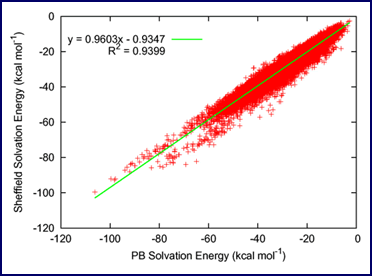
Accurate Poisson—Boltzmann vs. Sheffield Solvation Model solvation energies for a database of 64,000 drug-like molecules [3].