

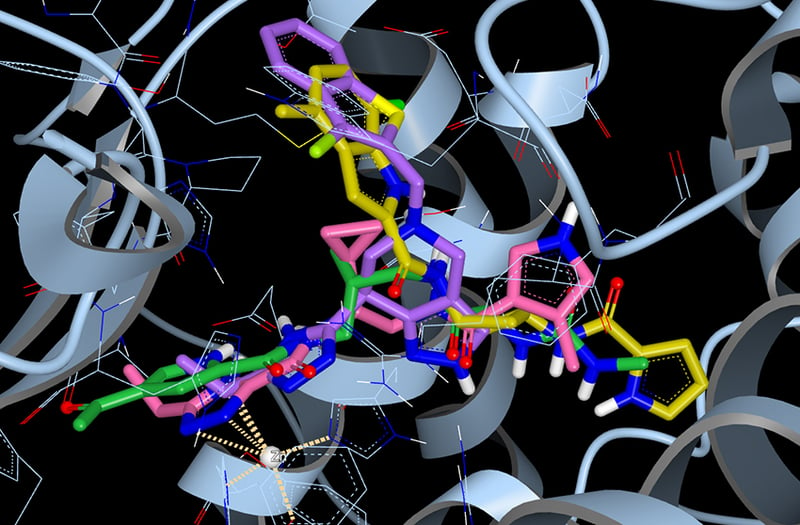
ACE2 with the top four scoring hits from our docking study. The four compounds were selected from the Enamine REAL library of approximately 1.4 billion synthesizable compounds. The top 10,000 hits from this docking study are freely available for download.
Summary: In an effort to help in the search to find potential therapeutics for COVID-19, OpenEye has completed multiple large-scale computational studies. The first dataset was released on April 27 (https://www.eyesopen.com/blog/openeye-deploys-the-orion-molecular-design-platform-to-find-covid-19-therapeutics). Here we publish a second such set of results and make them freely available to advance research toward a therapy.
COVID-19 is a pandemic disease which is caused by the novel coronavirus SARS-CoV-2. The disease causes fever, severe respiratory illness, and in some cases pneumonia. As of May 27, 2020, COVID-19 has resulted in 351,815 deaths globally1.
SARS-CoV-2 virus enters the host cell via attachment of its spike protein to the angiotensin converting enzyme 2 (ACE2) of the host cell. Structures of ACE2 bound to the spike protein of the SARS-CoV-2 virus (PDB IDs: 6M0J and 6LZG)2,3 indicate a protein-protein surface that facilitates spike attachment to ACE2. Examining ACE2 structural arrangement in the apo (PDB ID: 1R42)4 and inhibitor bound (PDB ID: 1R4L)4 states reveal a large conformational change upon ligand binding. Specifically, there is a remarkable structural rearrangement in the region of ACE2 that binds the spike protein. This suggests that ligand binding to ACE2 might disrupt the protein-protein surface necessary for viral entry into host cells. Therefore, ACE2 inhibition may represent a therapeutic opportunity for the treatment of COVID-19.
At OpenEye Scientific we sought to leverage our expertise, molecular modeling tools and the Orion molecular design platform for lead finding for potential treatment of COVID-19. We employed a molecular docking study at giga-scale which utilizes the three-dimensional structure of the target protein to identify lead compounds in large libraries (greater than 1 billion) of drug-like molecules.
Using the structure of ACE2 bound to an investigational drug compound, MLN-4760 (PDB ID: 1R4L), we undertook a giga-docking experiment to identify potential leads for ACE2 inhibition. In this experiment we screened the Enamine REAL compound library, a database of approximately 1.4 billion molecules that are designed to be readily synthesized. The Enamine REAL collection has been used successfully in docking experiments to identify new chemical matter in a variety of targets. The docking calculation to identify potential ACE2 inhibitors utilized OpenEye’s cloud-native molecular design platform, Orion, to search the approximately 266 billion conformers of 1.4 billion molecules and utilized 50,000 CPUs over 3 days at Amazon Web Services.
As a service to the larger scientific community and to help fight this pandemic, we are making the results available free of charge. In addition, we are seeking to connect with researchers to collaborate on testing further COVID-19 protein targets and molecules.
Please email info@eyesopen.com for details.
Data available for public use:
Hit list, scores and Enamine compound IDs
1R4L protein crystal structure prepared for docking
This work was supported by a generous grant from Amazon Web Services.
References
1. https://coronavirus.jhu.edu/map.html
2. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., Zhang, Q., Shi, X., Wang, Q., Zhang, L., Wang, X. (2020) Nature 581: 215–220.
3. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Wang, Q., Zhang, Y., Wu, L., Niu, S., Song, C., Zhang, Z., Lu, G., Qiao, C., Hu, Y., Yuen, K.Y., Wang, Q., Zhou, H., Yan, J., Qi, J. (2020) Cell 181: 894–904.
4. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. Towler, P., Staker, B., Prasad, S.G., Menon, S., Tang, J., Parsons, T., Ryan, D., Fisher, M., Williams, D., Dales, N.A., Patane, M.A., Pantoliano, M.W. (2004) J Biol Chem 279: 17996-18007.