Trusted Science. Delivered the Way You Need.
Large Scale Virtual Screening
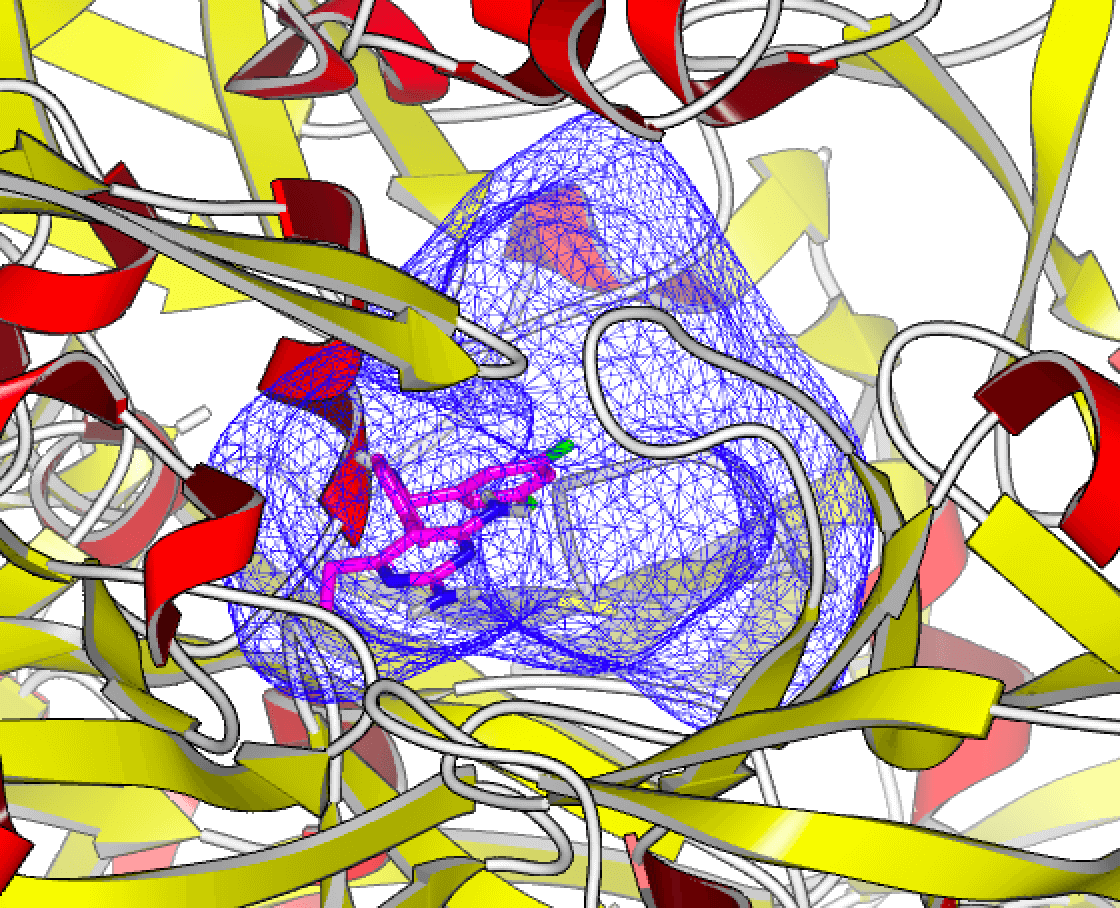
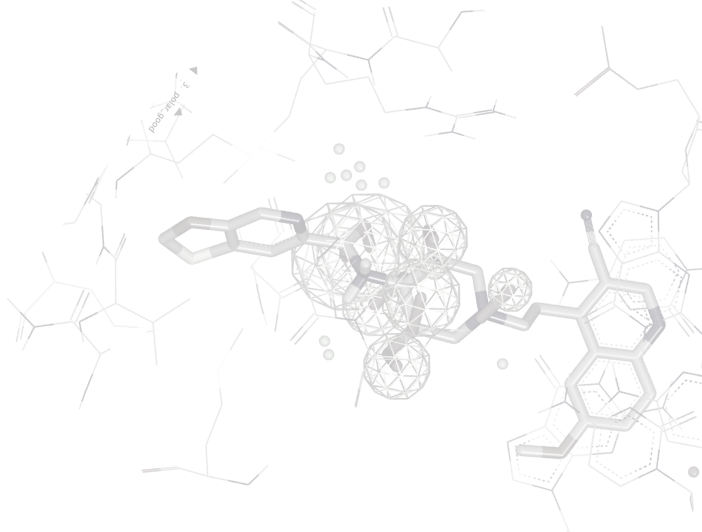
No matter the size of the space you want to search, OpenEye has a fast and cost-effective virtual screening option for you. Delivering 2D and 3D virtual screening solutions at the scale you need, the speed you want, and the computing cost you desire.
Sygnature Discovery relies on OpenEye's Orion for large-scale virtual screening (giga-scale docking) to quickly identify hits for their customers and scientists. Learn how.