

SPRUCE
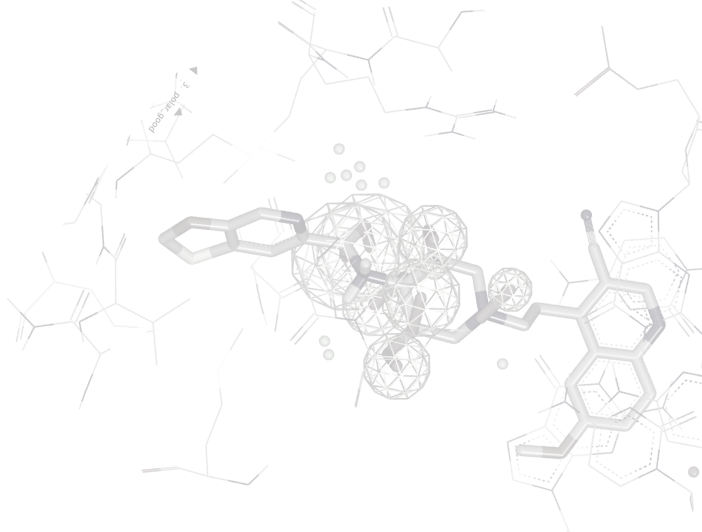
Reliable structure-based drug design begins with a structure that correctly represents the biology. SPRUCE transforms experimental data from PDB, mmCIF, or cryo-EM sources into fully prepared, biologically meaningful systems ready for structure-based drug discovery use such as in docking, binding free energy calculations, and molecular dynamics.
Errors introduced during structure preparation propagate through every downstream calculation. Misplaced hydrogens may corrupt docking scores and force field calculations. Incorrect protonation states distort binding pose predictions. Unmodeled loops leave binding site geometry incomplete. SPRUCE resolves these issues systematically, so your modeling results reflect real biology rather than preparation artifacts.
What SPRUCE Prepares and Why it Matters
Each preparation step addresses a specific source of error in downstream biophysical modeling.
- Hydrogen bond network optimization.
- Tautomer and protonation state enumeration.
- Biological unit assembly.
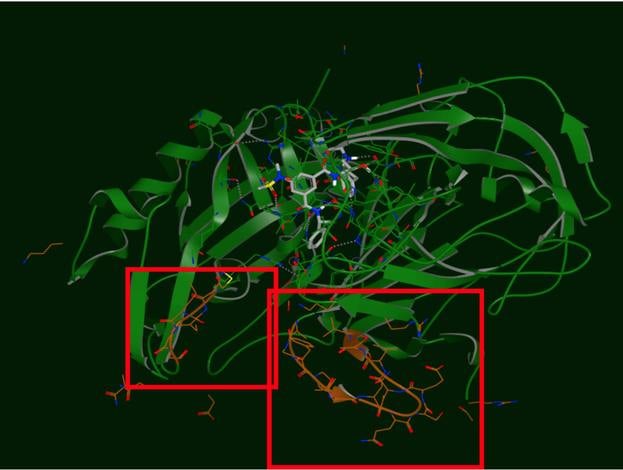
- Loop and missing residue modeling.
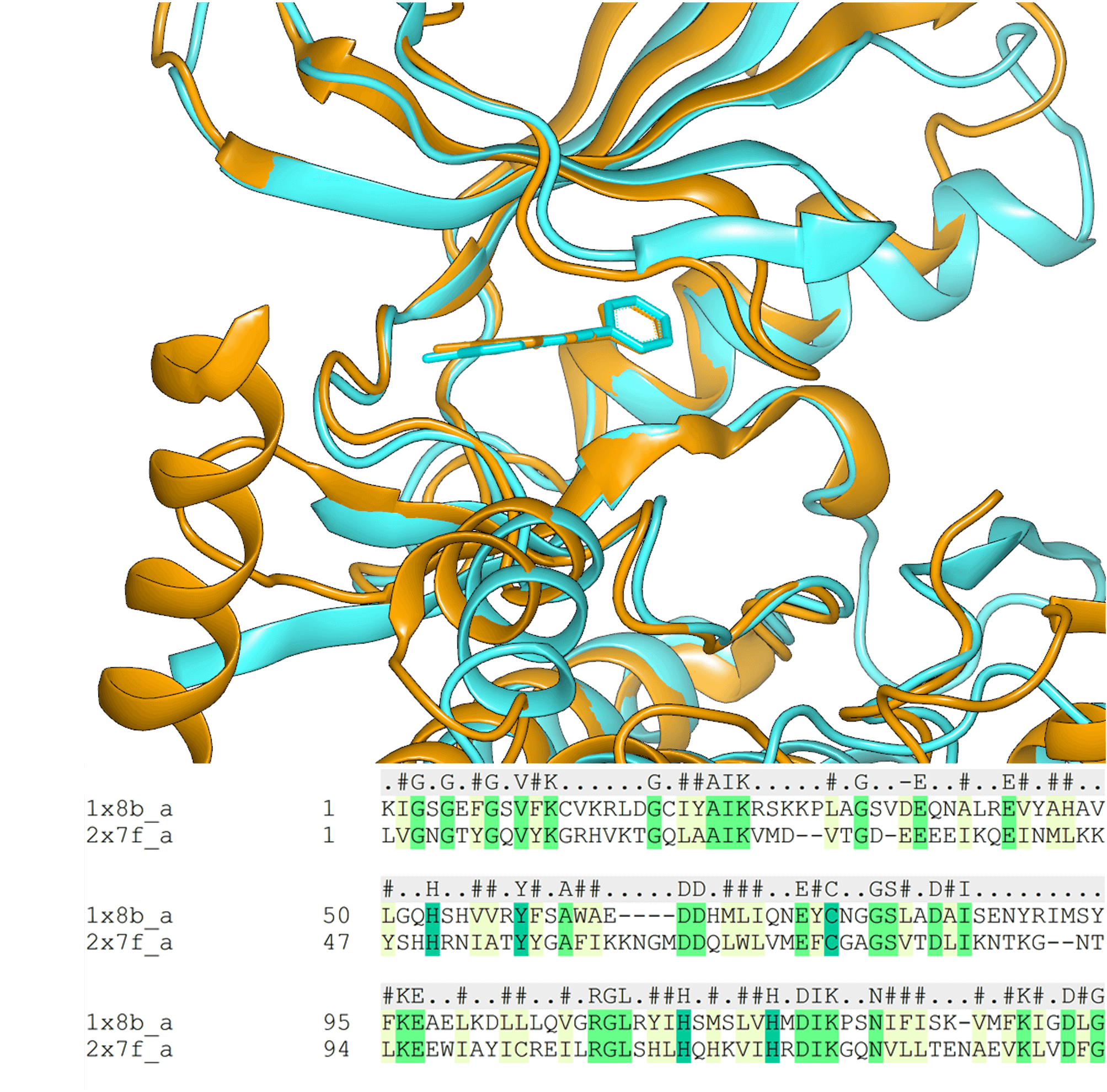
- Side-chain remodeling and point mutations.
- Enumerates pockets for apo structures.
- Protein superposition.
- Support PDB and mmCIF files.
How SPRUCE Works
SPRUCE streamlines the preparation process by automatically breaking down the system into individual biological components, adding any missing protons or residues, and subsequently optimizing the hydrogen bond network for the entire system.
SPRUCE's structure preparation workflow performs tasks including the enumeration of biological units, alternate locations (if present), modeling missing residues and loops, and placing and optimizing hydrogens, accounting for the likely tautomer states of bound heterogens (ligands and cofactors).


Structure Quality Assessment
Not all experimentally determined structures are equally suitable for modeling. Resolution, refinement quality, ligand fit to electron density, and crystal contacts all affect whether a structure will produce reliable predictions. SPRUCE integrates the Iridium classification system to give scientists a transparent, reproducible basis for selecting which structures to use in a campaign.
-
Iridium-HT: Highly trustworthy, suitable for direct use.
-
Iridium-MT: Moderately trustworthy, flagged issues present.
-
Iridium-NT: Not trustworthy, use with caution.
Structures flagged by Iridium receive detailed annotations identifying the specific issue, whether it is poor electron density, poor ligand fit to the density, or problematic crystal contacts. This allows scientists to select optimal protein structures or units for their drug discovery pursuit.

