

AFITT
AFITT is the only software to offer a fully automatic ligand fitting process that optimizes a real-space fit to density while keeping conformational strain to a minimum. It capitalizes on a combination of core technologies that OpenEye has developed, specifically conformer generation, shape potential, high-quality small molecule structure minimization, and visualization. The key step, after finding the appropriate conformers and aligning them to density, is the implementation of a refinement that combines force field and shape potentials, via a series of adiabatic optimizations [1, 2].
.png?width=623&height=371&name=AFITT_banner_pic1%20(2).png)
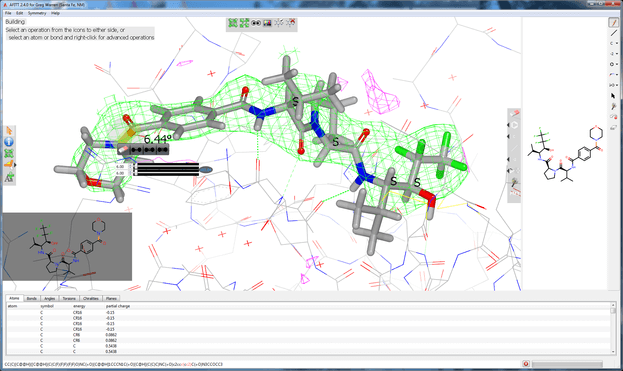
The AFITT command-line tools provide a powerful means for integration and automation, which is critical in the application of high-throughput crystallography for drug discovery.
For more detailed information on AFITT, check out the link below:
Documentation
