

COMPOUND PROPERTY CALCULATION & REMOVAL OF UNDESIRABLES
FILTER
FILTER is a very fast molecular filtering and selection application. It uses a combination of physical property calculations and functional group knowledge to remove undesirable compounds before they enter experimental or virtual screening.
Undesirable properties may include: toxic functionalities, a high likelihood of binding covalently with the target protein, interfering with the experimental assay, and/or a low probability of oral bioavailability.
Eliminating unwanted molecules before the use of modeling applications will, in turn, substantially increase the positive predictive value of these tools, and significantly reduce their processing time.
For more detailed information on FILTER, please click below:
Documentation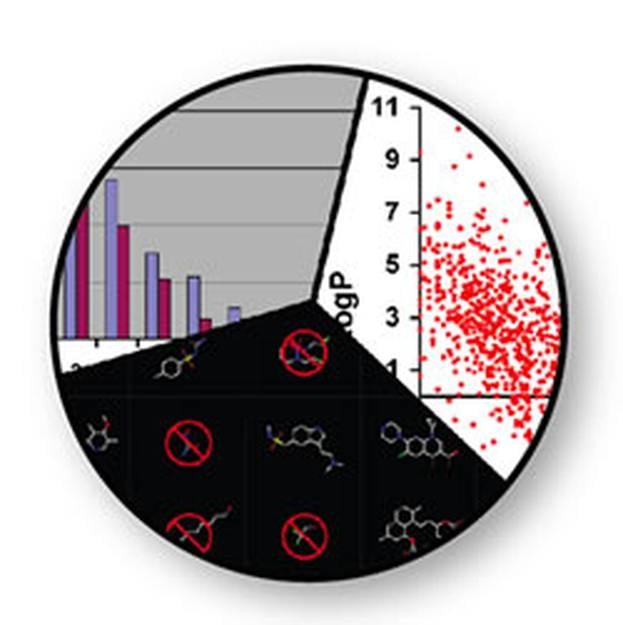
Removing undesirable compounds early makes the downstream processes significantly more efficient; a simple concept that is too often ignored.

