

ROCS®
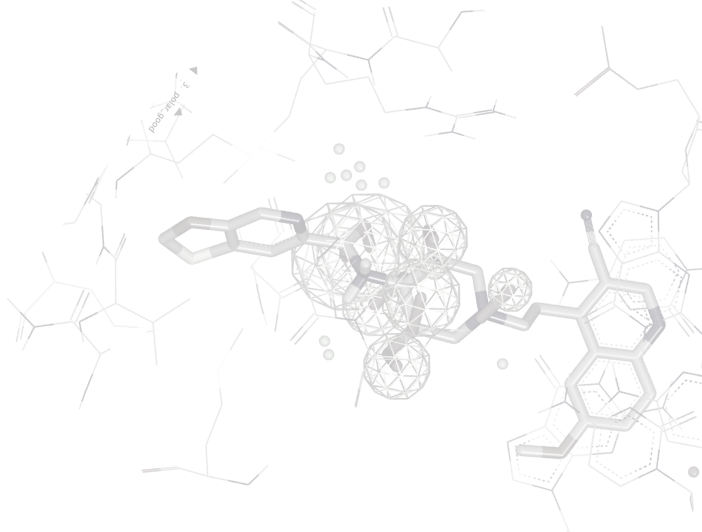
ROCS is powerful ligand-based lead discovery software that identifies potentially active leads by comparing molecules using shape and the distribution of chemical features, or color. Screening of databases can be carried out at hundreds of molecules per second on a single CPU, allowing rapid analysis of large collections of molecules. ROCS is competitive with, and often superior to, structure-based approaches in virtual screening [1,2], both in terms of overall performance and consistency [3]. Novel and interesting molecular scaffolds have been identified using ROCS against targets often considered very difficult for computational techniques to address [4].
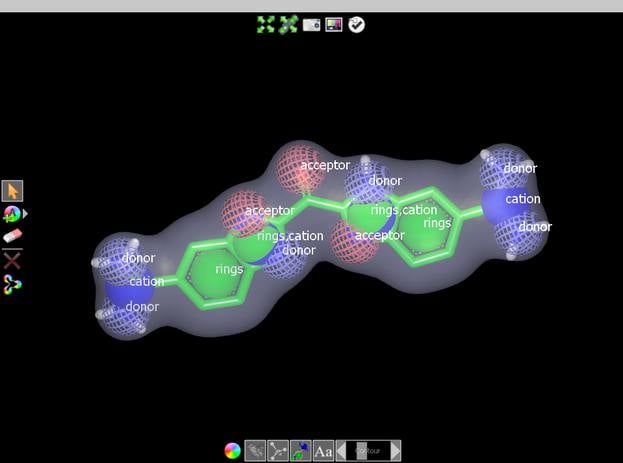
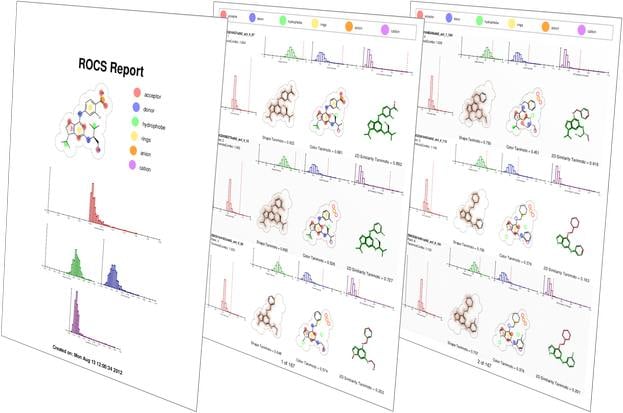
ROCS is the class-leading shape comparison application; it has been used successfully in hundreds of published studies to find new chemical matter with relevant biology. It uses a smooth Gaussian function to represent the molecular volume [5], so it is possible to routinely minimize to the best global match.
ROCS alignments have a number of applications: 3D-QSAR, SAR analysis, understanding of scaffold diversity and detection of common binding elements[6]. ROCS alignments to crystallographic conformations have also been useful in pose prediction in the absence of a protein structure [7].
vROCS is the graphical user interface for ROCS that enables users to jump right into working with ROCS. vROCS also provides a powerful query editor enabling the advanced user to design complex queries. Recognizing the importance of query validation, vROCS includes a collection of statistical tools to evaluate the performance of different queries.
For more detailed information on ROCS, check out the link below:
Documentation
