

OEDocking
OEDocking is a comprehensive suite of well-validated molecular docking tools and workflows, each tailored to tackle specific aspects of protein-ligand interactions. Whether you need fast exhaustive docking (FRED), ligand-guided docking (HYBRID), informed pose prediction (POSIT), or induced-fit posing (IFP), OpenEye offers the premier scientific tools for all your docking and virtual screening needs.
OEDocking is available as applications, cloud, and as a toolkit through OEDocking TK providing versatile options to suit your needs.
We identified the ureido methylpiperidine carboxylate derivative [using OEDocking], compound 7, as a reversible, selective, and potent inhibitor of cathepsin V. – Mitrovic et al. (CSBJ, 2022)
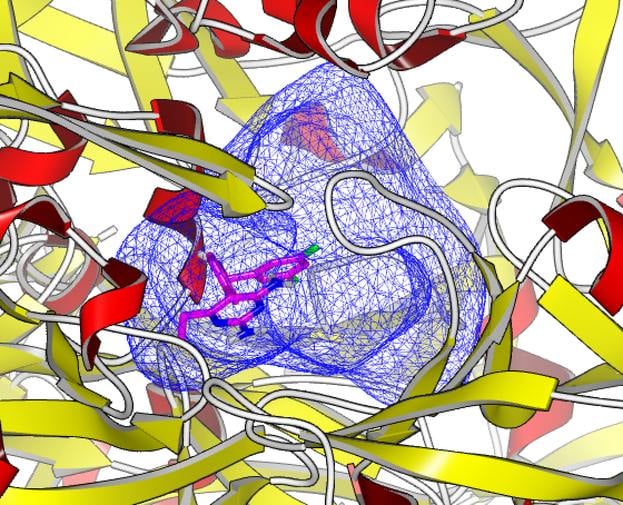
Docking and Virtual Screening
FRED - Fast Exhaustive Docking
Within a given, but practical, resolution FRED performs a systematic and non-stochastic examination of all possible protein-ligand poses, filters for shape complementarity and chemical feature alignment before selecting and optimizing poses using the Chemgauss4 scoring function [1,2,3,4]. It comes with a powerful GUI for preparing the active site and adding custom restraints. It also provides a detailed scoring analysis that uses our Grapheme toolkit. In one example, Brus et al used FRED to discover a validated 2.7nM inhibitor of BChE, an Alzheimer's target [5]. The authors describe FRED as "by far the fastest docking tool and thus particularly suitable for ultrahigh-throughput docking".
Especially useful when you possess information solely from the apo-protein structure.
HYBRID - Ligand Guided Docking
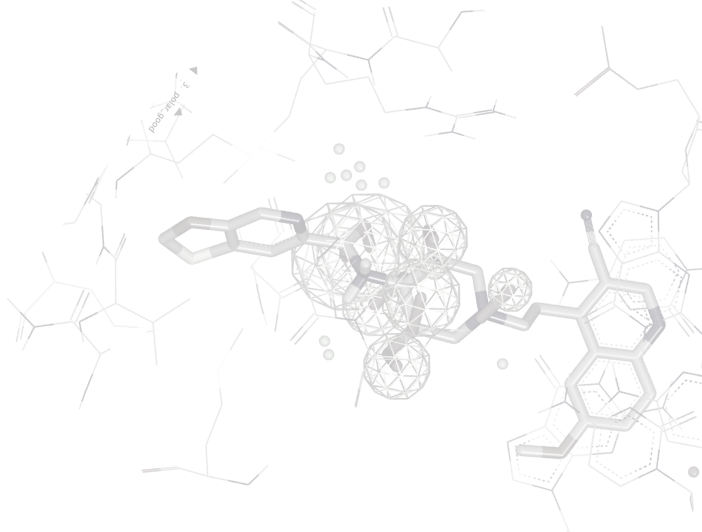
HYBRID uses bound ligand information to improve virtual screening performance, e.g. as POSIT improves poses HYBRID improves enrichment. Like FRED, HYBRID performs a systematic, exhaustive, non-stochastic examination of poses within the protein active site; however, HYBRID reduces this search space based on shape and chemical complementarity to the bound ligand. This ligand-guided docking provides equivalent or better enrichment when compared to most docking procedures [1,2].
Highly valuable when you have information from the holo-protein structure.
Gigadock™ - Ultra-Large Virtual Screening
Need to run docking on ultra-large compound libraries? Complementing our core OEDocking tools, Gigadock and Gigadock Warp offer 3D docking against tens of billions of compounds, providing the ultimate choice for the best in science, cost, and speed.
Ligand Posing and Analysis
POSIT - Knowledge guided pose prediction
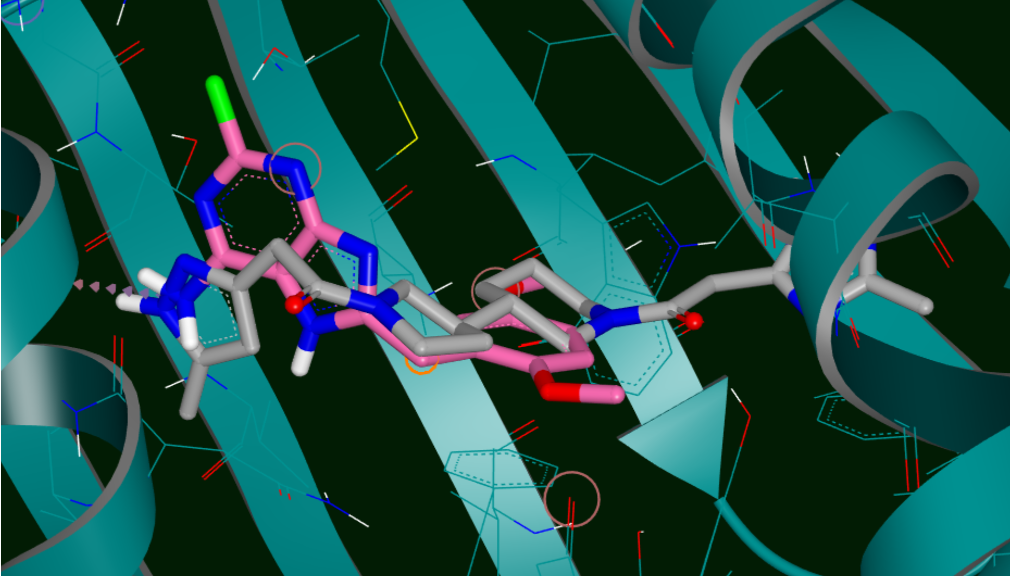
POSIT leverages bound ligand information for enhanced pose prediction. Using a combination of OpenEye's techniques, including structure generation, shape alignment, and flexible fitting, POSIT assesses a target ligand's similarity to bound ligands, guiding the algorithm and offering an accuracy estimate. Both 2D and 3D similarity measures are used in this reliability index, which is an industry first.
Furthermore, when given a set of ligand-receptor complexes from a crystallographic series, POSIT can autonomously identify the most suitable structure for guiding new ligand docking. The performance of this 'best guess' structure closely rivals that of using each structure individually and selecting the best result in hindsight. For single-pose generation, POSIT efficiently utilizes one structure, reducing computational load while maintaining high performance. For multiple pose generation, it offers the flexibility to use either the 'best guess' structure or all available structures for superior pose quality.
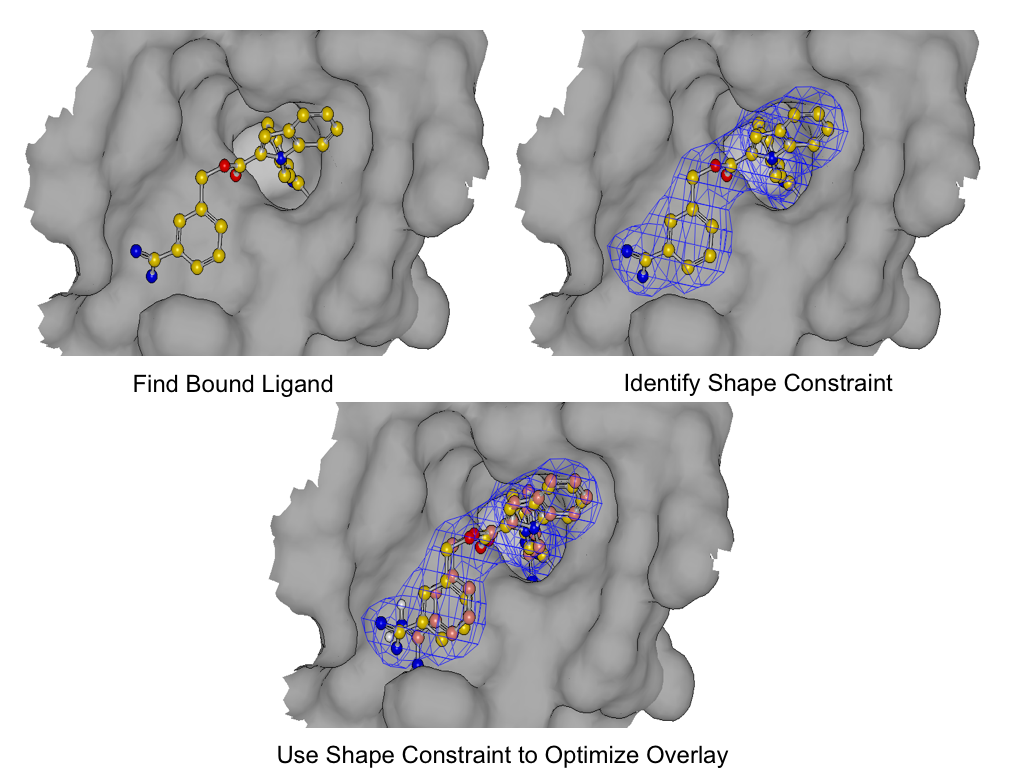
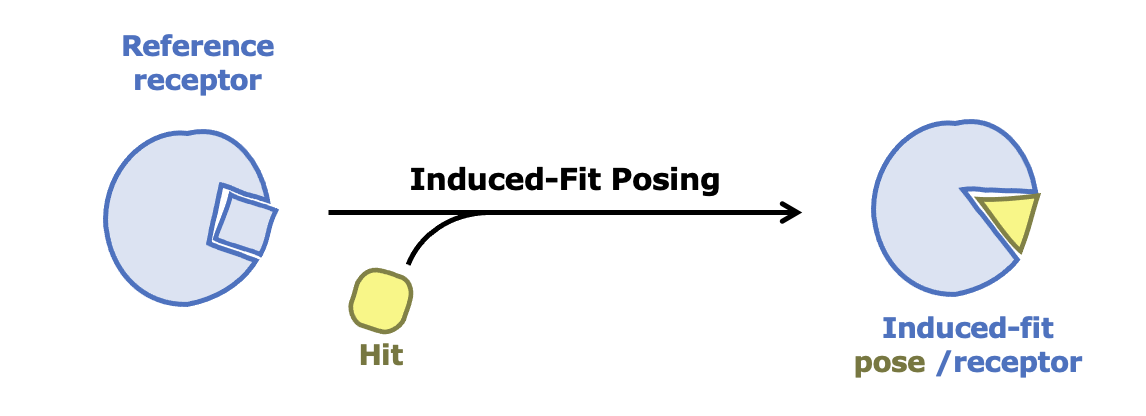
Induced-Fit Posing
To enhance your ability to predict ligand binding affinity for proteins with flexible target sites, consider leveraging both Induced-Fit Posing and Free Energy - Nonequilibrium Switching (FE-NES). These tools combined help provide more accurate predictions of ligand-protein binding.
Learn more about Induced-Fit Posing

