

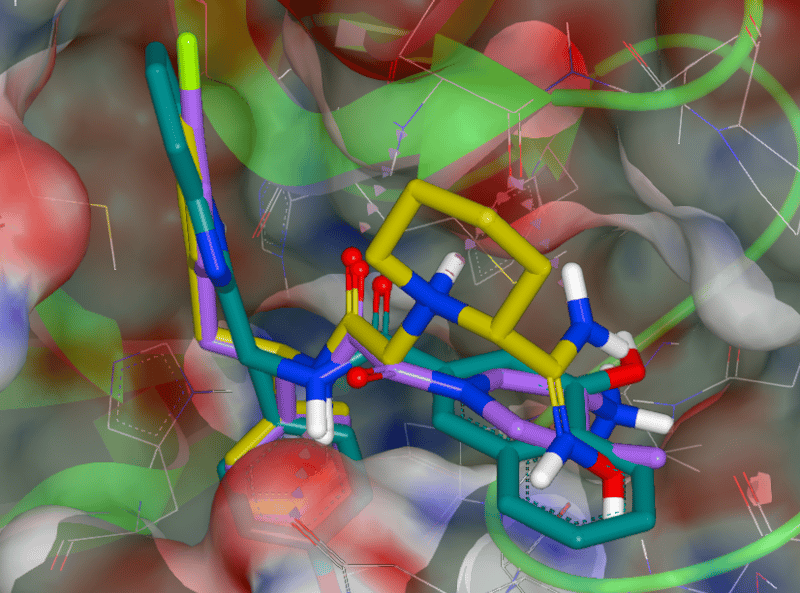
SARS-CoV-2 Mpro protease with the top three scoring hits from our docking study. The three compounds were selected from the Enamine REAL library of approximately 1.4 billion synthesizable compounds. The top 10,000 hits from this docking study are freely available for download.
Summary: In an effort to quickly find potential therapeutics for COVID-19, OpenEye has completed multiple large-scale computational studies. Here we publish the first such set of results and make them freely available to advance research toward a therapy.
The current pandemic of COVID-19 disease is caused by a coronavirus infection of a virus designated as SARS-CoV-2. Coronavirus treatments have been elusive, with clinical trials underway in an attempt to identify compounds to treat the infection. Targeting viral enzymatic function has been shown to be a successful approach in treating viral infections. There are several enzymes that are typically exploited in the viral genome, and one of these, the viral maturation proteases have been the focus of much work to develop anti-viral therapies.
At OpenEye Scientific we have had a long-standing interest in pharmaceutical lead finding by molecular docking, which utilizes the three-dimensional structure of the target protein to identify lead compounds in large libraries of drug-like molecules. Recently, we have extended this docking approach to very large libraries of compounds numbering in the billions that are designed to be readily synthesized. One such library, the Enamine REAL collection, has been used successfully in docking experiments to identify new chemical matter in a variety of targets. As of April 2020, the Enamine REAL library consists of approximately 1.4 billion molecules when enumerated properly in three dimensions.
We use the x-ray crystal structure of the protease from SARS-CoV-2 published by Liu, et al. (PDB accession number 6LU7). Using that structure we undertook a docking study to identify potential leads for protease inhibitors. The calculation utilized 50,000 CPUs and was enabled by Orion, OpenEye’s molecular design platform powered by Amazon Web Services (AWS). The calculation, which involved docking ~266 billion small molecule structures into the protein target, required 3.5 days to complete and was supported by a generous grant from AWS. As a service to the COVID-19 treatment development efforts, we are providing free access to the results of the calculations to enable testing of these potential therapies by researchers worldwide. In addition, we are seeking to connect with researchers to collaborate on testing further COVID-19 protein targets and molecules. Please email info@eyesopen.com for details.
Data available for public use:
Hits, scores, poses and Enamine compound IDs
6LU7 protein crystal structure prepared for docking
This work was supported by a generous grant from Amazon Web Services.