

By Gaetano Calabro PhD, OpenEye & Christopher Bayly, PhD OpenEye
A variety of free energy simulation methods, such as FEP, TI, and λ dynamics, make use of atomistic MD or Monte Carlo simulations to determine the relative binding free energy (RBFE) between two ligands along an alchemical path. Non-Equilibrium switching (NES) is a relatively new method to estimate binding affinities based on the calculation of thermodynamic work along fast, non-equilibrium alchemical paths. In Orion, an implementation of De Groot’s RBFE-NES method has been tested on few targets.
Introduction
RBFE calculations are increasingly used in pharmaceutical lead optimization to get a quantitative estimate of the effect of inhibitor changes on affinity. By alchemically morphing a known ligand into a hypothetical candidate this difference can be assessed directly, taking advantage of similarities between the two inhibitors.
NES for RBFE has been implemented in Orion mainly using the current MD Orion infrastructure. Alchemical mutations between selected ligands have been implemented using de Groot’s PMX package(1) to create the "chimera" molecule between the morphing ligands.
In the NES protocol, after equilibrium simulations, sets of phase states are selected as initial conditions to run fast MD Non-Equilibrium transitions, evaluating thermodynamic work to mutate a ligand into another and vice as shown in Figure 1.
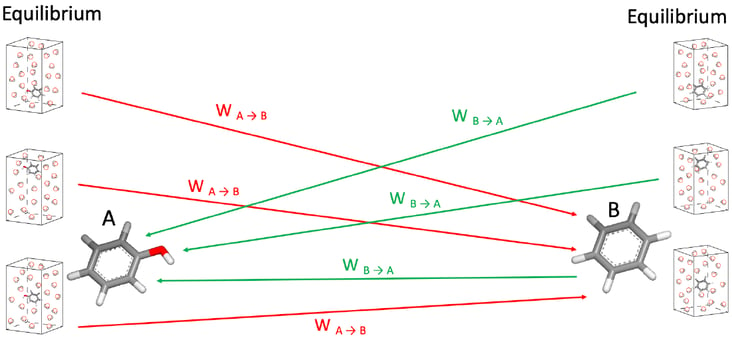
Figure 1: Snapshots from equilibrium MD runs are selected as initial conditions to run NES calculations. In the picture the ligand A in solution (Unbound State) is quickly alchemically mutated into the ligand B (and vice versa); the work is tracked to estimate
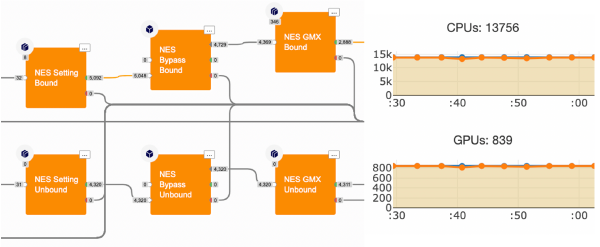
The NES protocol particularly suits the Orion infrastructure. Indeed, each non-equilibrium alchemical mutation is an independent MD simulation. Thousands of RBFEs can be calculated simultaneously, benefiting from Orion’s large scaling capability. See Figure 2.
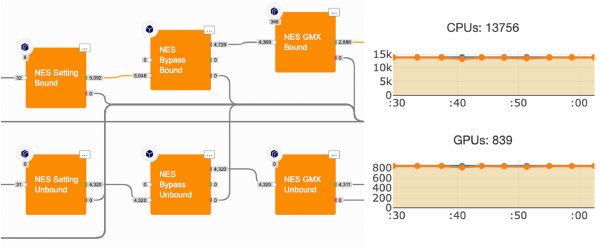
Figure 2: In Orion, thousands of fast MD runs are performed in parallel to calculate the work to alchemically mutate a ligand into another.
Results
Two targets, Thrombin and CDK2, and their inhibitors were selected to start testing the NES implementation in Orion. Figure 3, 4 shows what is produced in Orion for Thrombin and Figure 5 for CDK2..
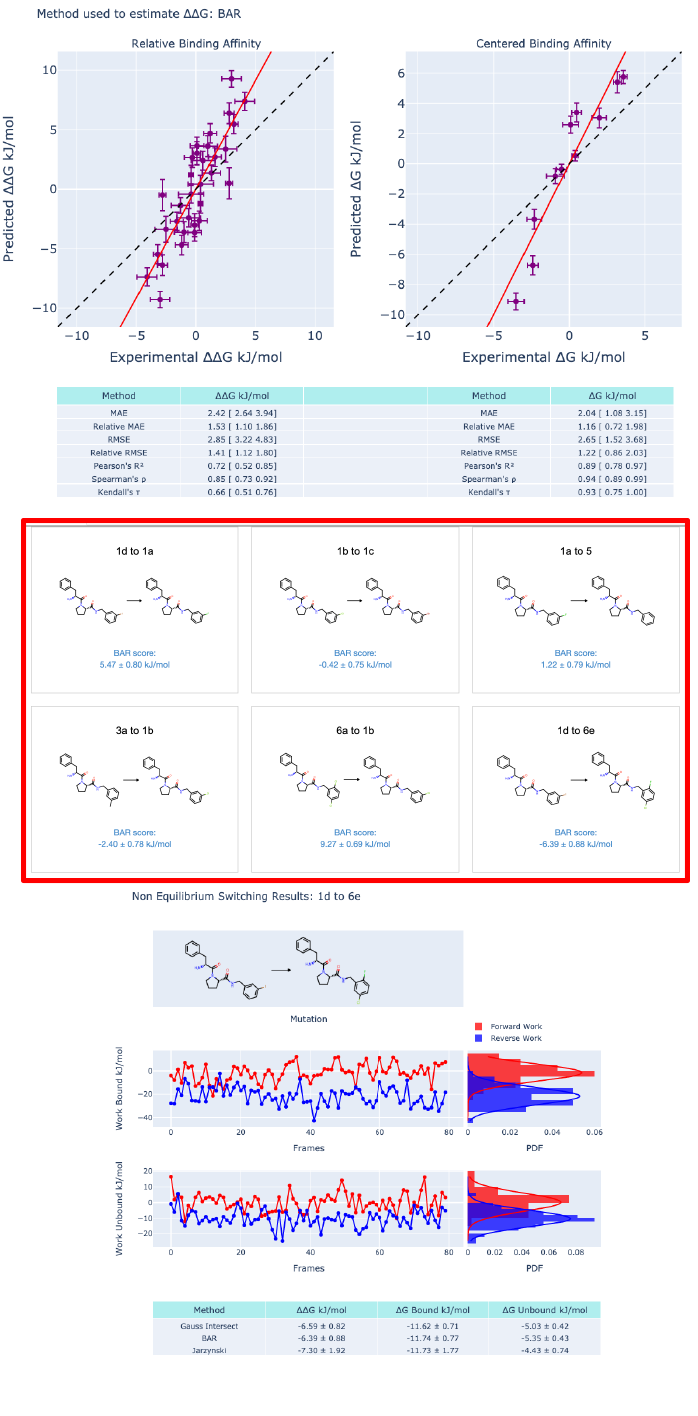
Figure 3: Results for the Thrombin target (protein 2ZFF prepared using Spruce). 11 ligands for 16 RBFE edges have been simulated in Orion. Each NES calculation used 80 frames and 50ps running time as in ref. 2.
To validate the NES implementation in Orion, Thrombin and CDK2 targets and the related ligands posed in the target binding sites were selected based on the Gapsys’ et al. publication(2,3)
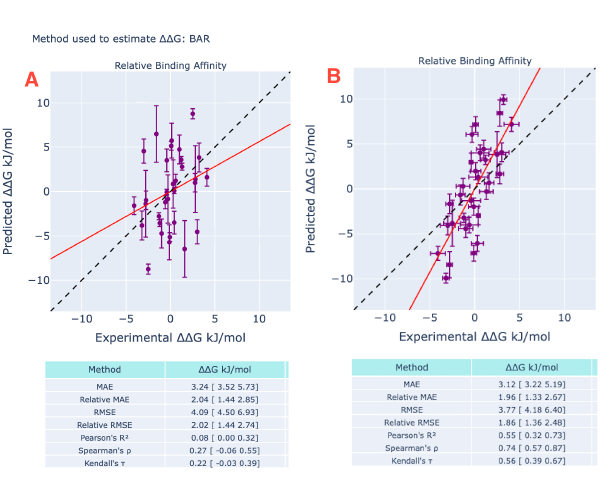
Figure 4: Thrombin RBFE results. In (A) symmetrized(4) ∆∆G evaluated from the de Gapsys’s publication(2,3) and in (B) the Orion results. One of the main differences between the runs is the selected protein forcefield. In (A) amber99SB*ILDN was selected while amber14 in (B). Ligands were parametrized by using Gaff2; ligand charges are am1-bcc in (A) while am1-bcc ELF10 charges were used in (B).
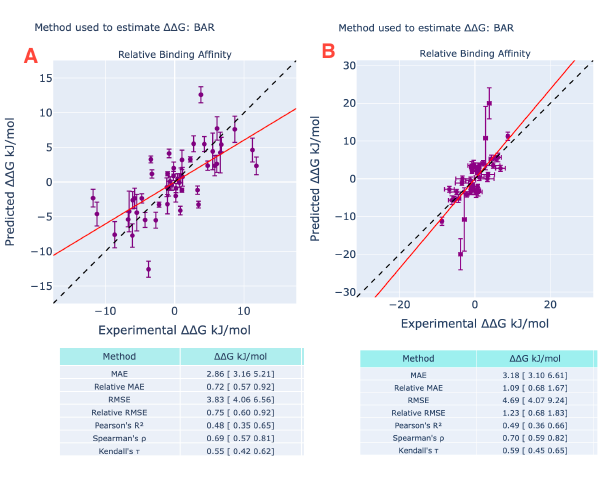
Figure 5: CDK2 RBFE results. In (A) symmetrized(2) ∆∆G evaluated from the de Gapsys’s publication(1,2) and in (B) the Orion results. The main differences between the runs are the same as reported in Figure 3. In Orion significant outliers were recorder and 5/25 edges were lost during the bound minimization stage.
Conclusions
In Orion, a starting implementation of the Non-Equilibrium Switching method has been implemented and tests are in progress. The NES methods to estimate RBFE suits Orion very well and its scaling capability to run thousands of fast MD runs in parallel. Initial results are encouraging but further development and validation is needed.
Acknowledgments
Andrew Shewmaker, Forrest York, Jayanth Shankar, Quinn Bailey, Scott White, Daniel Garrison, James Haigh.
David Mobley, John Chodera, David Hahn, Hannah Bruce Macdonald
References
(1)Gapsys V. et. al. (2014). pmx: Automated protein structure and topology generation for alchemical perturbations. The Authors Journal of Computational Chemistry. 36, 348-354
(2)Gapsys V. et. al. (2020). Large Scale Relative Protein Ligand Binding Affinities Using Non-Equilibrium Alchemy. Chemical Science. 11, 1140-1152
(3)David Hanh https://github.com/openforcefield/PLBenchmarks
(4)Hannah Bruce Macdonald, David Hanh https://github.com/choderalab/freeenergyframework