




SCIENCE
Use physics-based design to advance
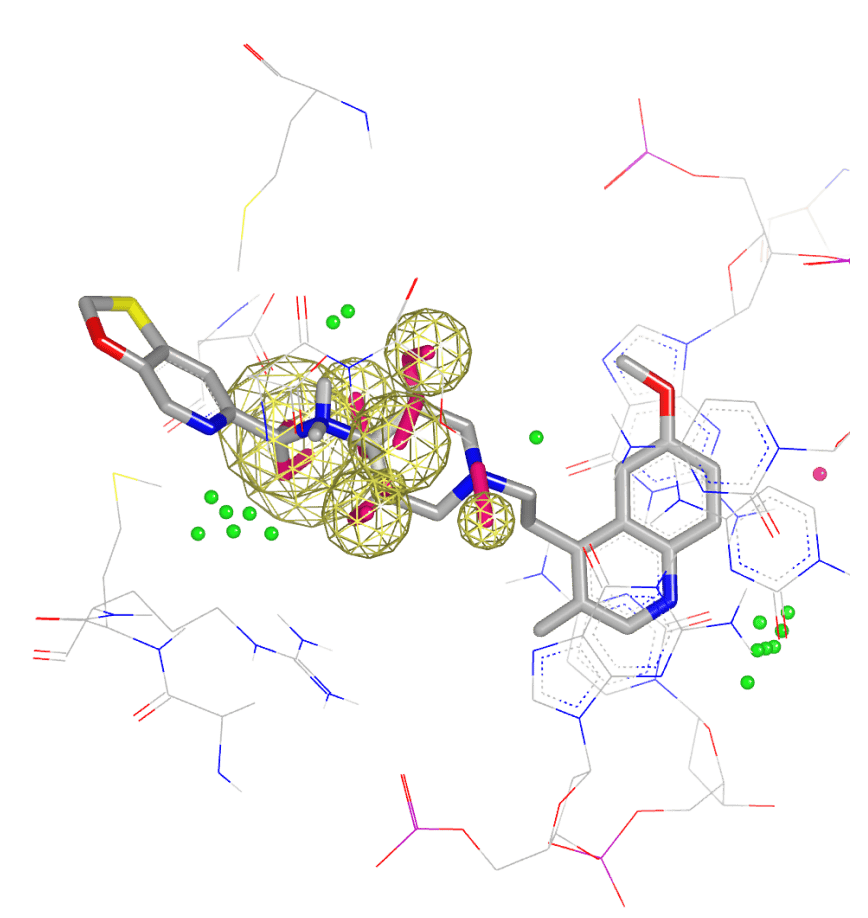
- Biomolecular target exploration
- Hit identification, hit-to-lead, and lead optimization
- Free energy predictions
- Pharmaceutical formulations

SCALE
Accelerate science to the speed of now
- Screen ultra-large scale compound databases
- Perform long timescale MD simulations on large systems
- Improve impact using rigorous theory & efficient algorithms
- Design & screen antibody libraries using NGS

